
期刊:Science China-Life Sciences
影响因子:8.0
scRNA-seq 的原始数据格式和目前大多数scRNA-seq 分析过程都基于 FASTQ 文件(或压缩格式 fq.gz)。Illumina 平台测序数据默认生成 BCL 格式文件,可以通过 CellRanger mkfastq 进行转换。scRNAseq的分析流程包括数据预处理、处理和扩展下游分析(图4),其中数据预处理包括质控、read比对和表达量化;数据处理包括标准化、批次效应校正、归一化、特征选择(HVG选择)、降维与聚类、细胞分型注释、差异表达分析(DEGs)、可视化;扩展下游分析包括拟时序、细胞间相互作用(CCI)、通路富集分析、基因调控网络(GRN)等下游分析。整体来看,scRNAseq分析方法层出不穷,没有绝对完美适用于所有场景的方法,分析工具重要的是获取生物学信息,难点在于选择最合适的方法。本文中,我们将提出总结常见的单细胞转录组分析方法,并对其优缺点和适用范围提出建议。
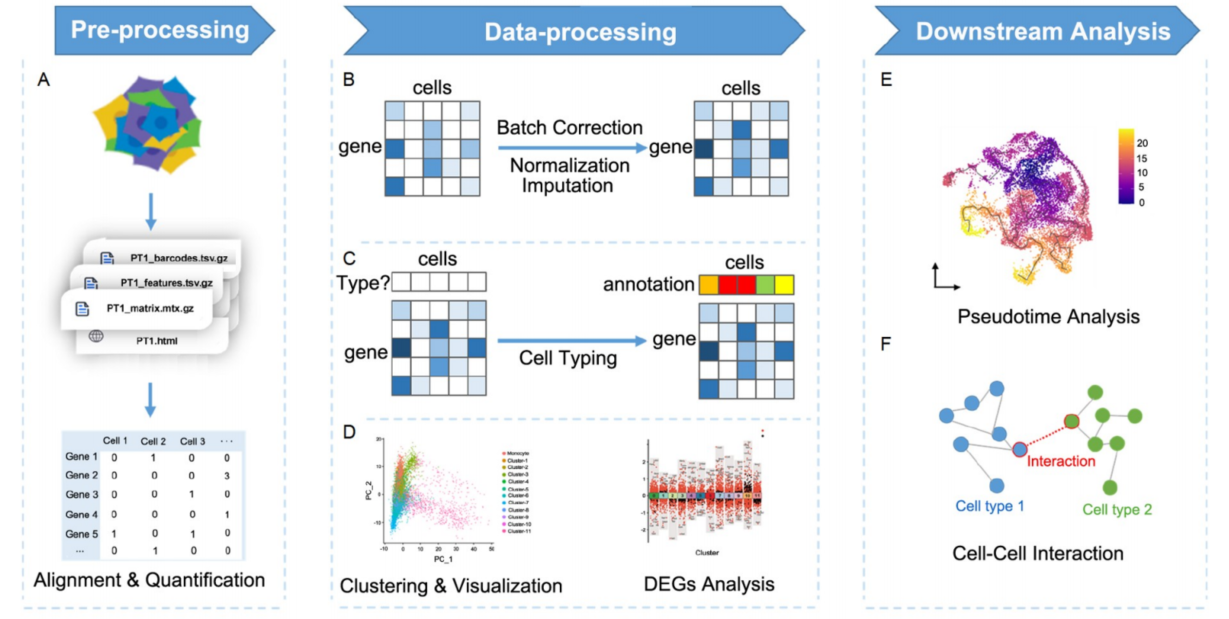
图4:单细胞分析概览。A. 在预处理阶段,基于测序数据,细胞-基因矩阵读数通过单细胞读数校正和定量产生;B. 分析使用的高质量细胞矩阵通过原始的基因表达矩阵获得,通过去批次效应矫正批次,通过标准化降低生物学差异,补充未检测到的基因;C. 依照或不依照先前的参考信息对细胞类型进行注释;D. 转录组特征相似的细胞被归为一类,称为“细胞簇(cluster)”,细胞的可视化通过降维方法实现,差异基因分析对组间差异进行检验;E. 拟时序分析重建细胞转录水平变化的动力学过程;F. 细胞间转录组调控关系可以通过胞间互作分析进行推断。
数据预处理
将原始测序数据通过滤除低质量reads和环境干扰与参考基因组进行比对和量化。从而得到每个细胞的特征计数矩阵和记录其他信息的辅助文件,用于下游的数据分析(图4 A)。
(1)质控
由于测序仪器问题、人为操作、细胞自发情况,或存在空液滴、双细胞、死细胞等,不可避免地会产生低质量的测序数据(Chen等,2019a ; Hao等,2021b)。空液滴通常出现在液滴捕获细胞外背景转录本而不是细胞时(Ilicic等,2016 ; Kolodziejczyk等,2015)。一种高度主观的方法是根据曲线的膝点确定一个UMI阈值,并过滤掉UMI计数低的细胞。随后使用DropEst ( Petukhov等,2018 )、EmptyDrops ( Lun等,2019 )和 DIEM ( Alvarez等,2020 )增强过滤效果。DropletQC ( Muskovic and Powell, 2021 )量化未剪接前 mRNA 含量的核分数得分。MT 基因阈值虽然是衡量死细胞的标准,但它的选择需要综合考虑细胞生理因素 ( Subramanian等,2022 )。近年来,基于深度学习的方法也应运而生,例如基于神经网络的 EmptyNN ( Yan等,2021 )和基于深度生成模型的 CellBender ( Fleming等,2019 ),能够有效识别空液滴中的背景转录本。
双细胞是指两个细胞包含在一个液滴中的情况,根据转录分布可分为同源双峰和异源双峰,均服从泊松统计量(Bloom, 2018)。绝大多数方法基于基因表达计算,利用先验知识或深度学习获取单峰与双峰细胞的差异,然后训练分类器进行筛选,例如基于最近邻的 DoubletFinder ( McGinnis等,2019a )、Scrublet ( Wolock等,2019 );基于反卷积的 DoubletDecon ( DePasquale等,2019 )、基于变分自编码器的 Solo ( Bernstein等,2020 )和基于集成算法的 Chord ( Xiong等,2021a )。此外,Scds 是另一种筛选方法,它依赖于基于共表达的双联体打分和基于二分类的双联体打分策略,实现 scRNA-seq 表达数据的双联体分离 (Bais and Kostka,2020)。一些方法使用其他特征,例如 demuxlet 它使用自然遗传变异信息指导实验并通过计算进行过滤 ( Kang等,2018 )。
合理的质控需要综合考虑技术性和生物性因素,这也是当前研究的主要方向。最近一种由生物数据驱动的自学习无监督质控方法 ddqc被提出来,用于确定各种 GC 指标的具体阈值 ( Macnair and Robinson,2023 )。
(2)reads比对和定量
质控后剩余的高质量细胞需要将这些短reads映射到特定的参考基因组上进行比对,以此对基因表达水平进行定量。RNA比对通常分为两步:比对reads以建立索引和映射RNA剪接序列,前一步与DNA reads比对共用,解决错配问题并设置索引参考;后一步是RNA reads比对所特有的,提供连通性信息。
早期二代测序结果是几十对长度的碱基reads。Seed-to-extend ( Buhler,2001 )(包括MAQ ( Li等,2008a )、SOAP ( Li等,2008b )、CloudBurst ( Schatz,2009 )、ZOOM ( Lin等,2008 ))、BurrowsWheeler 变换方法 ( Burrows and Wheeler,1994 )(包括SOAP2 ( Li等,2009 )、Bowtie ( Langmead等,2009 )、BWA ( Li and Durbin,2009 ))、Needleman-Wunsch 方法(包括Novocraft ( Hercus,2009 ))和suffix-tree算法方法(包括MUMmer 2 ( Delcher等,2002 ))都是百万级短链 DNA 测序reads比对的有效工具。Bowtie采用了一种依赖于Burrows-Wheeler Transforming的FM-index方法,如果reads有多个准确匹配则结果只报告一个,与MAQ(Ferragina and Manzini, 2001)相比,大大优化了运行内存和比对速度。BWA是另一种基于BWT的比对方法,使用新的SAM(Sequence Alignment/Map)格式输出比对结果。基于MAQ和Bowtie两种短链DNA比对算法,Cole Trapnell于2009年提出了第一个针对NGS数据的RNA-seq比对方法TopHat,它使用2-bit-per-base编码实现reads与哺乳动物基因组中剪接位点的有效比对,而无需事先知道剪接位点的具体信息(Trapnell等, 2009)。
上述方法在碱基对长度超过50 bp时比对精度急剧下降(Gupta等,2018 ; Lebrigand等,2020)。NGS单细胞测序分析主要采用两类方法:基于Bowtie2的方法和基于seed策略的方法(Langmead and Salzberg, 2012)。Bowtie2是Bowtie的升级版,保留了FM-index依赖的BWT算法核心,允许有间隙比对,并使用单指令多数据(SIMD)扩展到长测序比对,同时提高运行速度。Daehwan Kim在Bowtie2基础上,先后提出了TopHat2(Kim等,2013)和HISAT(Kim等,2015)。种子策略主要有STAR(Dobin等,2013)和Subread(Liao等,2013)。STAR基于最大可映射前缀(MMP)的思想,采用顺序检索的策略,将与参考匹配的最长部分reads设为种子1,其余read继续匹配,依次从种子2调用至种子n 。值得注意的是,Rsubread完全基于R语言平台实现了第一次read比对和基因量化的过程(Liao等,2019)。
基因表达量化又可分为伪比对量化和基于read比对的量化。伪比对是指不采用上述严格的两步法将所有reads比对到参考基因组上,包括选定的 k-mers 比对方法(Sailfish(Patro等,2014)、Kallisto(Bray等,2016)、Salmon(Patro等,2017)、RapMap(Srivastava等,2016))和 Barcode-UMI-Set (BUS)比对方法 BUStools(Melsted等,2019)。Kallisto-BUStools 是最新的工作流程,它使用 BUS 文件格式进行初始数据预处理,与 BUStools 一样,伪比对结果和量化计数都保存在 BUS 文件中(Melsted等,2021)。另一方面,基于reads比对的方法依赖于 RNA reads比对方法的结果来量化基因。CellRanger 是10x Genomic 公司指定替代 Longranger 的官方开源数据预处理软件(Zheng等,2017)。STARsolo 是替代 Cellranger 的mapping/quantification 功能的工具,可实现多平台测序数据的分析和基因表达之外的转录组特征的量化(Kaminow等,2021)。其他基于reads比对的基因表达定量方法如 UMItools ( Smith等,2017 )、zUMIs ( Parekh等,2018 )、Alevin-fry ( He等,2022 )、DropEst ( Petukhov等,2018 )、RainDrop ( Niebler等,2020 )、baredSC ( Lopez-Delisle and Delisle, 2022 )、BCseq ( Chen and Zheng, 2018 )使用各种质量过滤器和 barcode/UMI 处理策略在一定程度上提高了 CellRanger 的性能。
CellRanger 和 STARsolo 在处理包括 10x Chromium 在内的各种单细胞转录组数据集时都具有良好的运行速度,并且准确度极高。但在获得几乎相同结果的前提下,后者相比前者提升了至少5倍的运行速度,这也验证了Alexander Dobin等人使用STARsolo取代CellRanger的目的(Brüning等,2022 ; Chen等,2021a ; You等,2021)。
数据处理
在对表达矩阵进行必要的调整(Normalization、Batch Effect Correction、Imputation)后,即可从单细胞转录组数据中充分挖掘出生物信息进行分析。Seurat和Scanpy分别基于R和Python对上述过程进行模块化、可扩展的处理,是目前单细胞转录组数据的主流分析流程(Satija等,2015 ; Wolf等,2018)。常规分析流程和预期处理结果可参见总分析框架(图4 B-D)。
(1)标准化
在测序过程中,技术原因或者细胞本身的生物学差异可能造成同一样本内(细胞之间)或者不同样本之间的文库大小差异(Marinov等,2014)。无限数方法按照文库大小进行处理,按照具体原理大致可以分为基于全局缩放的标准化、spike-in标准化和其他数据变换模型标准化。
全局缩放方法最初是为bulk RNA分析而发展起来的,通过特定的缩放因子对全局数据进行缩放(Finak等,2015)。每万计数(CPT)变换和每百万计数(CPM)变换是常见的线性缩放方法,在不考虑spike-in count的情况下,它们都对每个UMI/总UMI count等距缩放。其他标准化方法包括每百万reads数(RPM)(Mortazavi等,2008)、修剪均值M值(TMM)、DESeq(Robinson and Oshlack, 2010)、上四分位缩放(Bullard等,2010)、FPKM(Trapnell等,2010)、RPKM(Tu等,2012)等,它们对于极值的稳定性比线性缩放更好,因此与RPKM/FPKM一样具有更广泛的应用范围。但单独使用该类方法进行单细胞转录组的标准化时,由于数据的稀疏性和假阳性率虚高,效果并不可接受(Evans等,2018),与特定方法结合时往往需要改进。SCnorm使用分位数回归方法来评估不同测序深度依赖细胞组之间的尺度因子(Bacher等,2017)。bayNorm基于基因原始计数与真实计数服从负二项(NB)分布的假设,使用集成贝叶斯模型对scRNA-seq数据进行标准化(Tang等,2020)。
spike-in标准化方法可以看作是全局尺度方法的另一种扩展,因为尺度因子是根据spike-in基因计算出来的。需要注意的是,将RNA spike-ins的信息添加到其他方法中也可以提高SCnorm等标准化的效果。GRM是一种基于spike-in ERCC分子浓度伽马分布的方法,其中ERCC是测序中常用的校准材料(Ding等,2015)。BASiCS 是一种自动贝叶斯标准化方法,将泊松分层模型应用于spike-in(技术)基因,以推断细胞特定的标准化常数(Vallejos等,2015)。
以上方法都是在细胞内RNA数量恒定的假设下对基因进行缩放,而这可能具有欺骗性,因此其他转化模型采用了不同的策略。由于单细胞转录组数据存在零膨胀问题,一些模型就是为此而设计的,例如相对对数表达(RLE)方法ascend(Senabouth等,2019)和基于NB的模型,如Dino(Brown等,2021)、scTransform(Hafemeister and Satija, 2019)。其他转化模型归一化方法如MUREN使用最小二乘(LTS)回归算法(Feng and Li, 2021);Sanity使用从UMI计数推断出的对数转录商(LTQ)作为贝叶斯框架的输入,以避免泊松波动,因为LTQ向量的变化估计了基因表达值(Breda等,2021);PsiNorm 是一种基于无监督帕累托分布尺度参数的方法,用于提升标准化效率和准确率(Borella等,2021)。Charles Wang 比较了 sctransform、TMM、DESeq 等共 8 种标准化方法,其中 sctransform 和 logCPM(Seurat 的内置处理方法)受数据影响最小,在可变数据集上最稳定(Chen等,2021a)。
(2)批次效应校正
由于实验设计、测序平台、测序时间、人员操作流程等原因,不同的单细胞转录组测序数据在mRNA捕获效率、测序深度等会存在明显差异,从而产生样本间的批次效应(Chen等,2019a ;Hwang等,2018 ;Tung等,2017)。理论上可以通过实验策略消除技术变异,但由于实验过程的客观限制以及测序仪器误差,不可避免地会或多或少地引入批次效应。利用计算方法进行校正是解决不完善实验设计的必要手段,通常使用的方法可以分为相互最近邻(MNN)方法、基于潜在空间的方法、基于图的方法、DL方法和其他方法。
MNN首先识别出不同批次之间同一细胞类型的最相似细胞,然后利用这些细胞进行批次效应校正,包括batchelor(Haghverdi等,2018)、Scanorama(Hie等,2019)、Canek(Loza等,2022)。另一类使用 MNN 的方法是基于降维后的潜在空间,如 Seurat ( Satija等,2015 )、BEER ( Zhang等,2019b )、SMNN ( Yang等,2021a )、iSMNN ( Yang等,2021b )。例如,Seurat 使用典型相关分析 (CCA)潜在空间中的 MNN 对 (称为“锚点” )来匹配相似细胞,而 BEER 使用主成分分析 (PCA)子空间来筛选相似性较差的子群。SMNN 和 iSMNN 分别采用监督机器学习和迭代监督机器学习来细化从预校正细胞聚类或迭代细胞聚类信息中训练出的MN对。
基于潜在空间的方法是指在隐藏空间或降维后的嵌入中进行批次效应校正的方法,除了基于 MNN 聚类的策略外,还有与 PCA 相关的空间方法 Harmony(Korsunsky等,2019)、FIRM(Ming等,2022)、Monet(Wagner, 2020);t 分布随机邻域嵌入 (tSNE)空间方法 sc_tSNE(Aliverti等,2020)和 ZINBWaVE(Gao等,2019)。Harmony 被广泛用于去除样本间的批次效应,使用 PCA 方法将排序的细胞输入到单个公共嵌入中,然后在最大多样性聚类和线性批次校正之间迭代循环,直到为每个细胞分配一个特定的校正因子,可用于后续的批次效应去除。Sc_tSNE方法引入梯度下降算法对传统t-SNE算法进行优化,随后采用线性校正(Aliverti等,2021)。ZINB-WaVE最初设计用于在单细胞数据中进行基因提取, Risso et al.(2018)将该方法扩展至小批量优化。
基于图的方法利用细胞基因表达矩阵将数字信息转化为空间构造的图,其中节点代表不同类型的批次,边的权重基于不同的计算方法。BBKNN利用k近邻细胞构建图(KNN图),通过使用均匀流形近似与投影(UMAP)方法合并不同数据集间单个细胞的图实现批次效应校正,这也是Scanpy工作流程中的默认方法(Pola ński等,2020 ; Wolf等,2018)。王波在 OCAT 中提出“幽灵细胞” (默认为 k-means 算法聚类中心)来制作细胞连接的二分图(Wang等,2022a)。
近年来,深度学习方法的快速发展也为批次效应校正提供了新思路,实现高效、大通量的数据处理,如 INSCT(Simon等,2021)(三重态神经网络)、CLEAR(Han等,2022)(自监督对比学习)、BERMUDA(Wang等,2019e)(迁移学习)、iMAP(Wang等,2021a)(VAE-GAN)、ResPAN(Wang等,2022e)(Wasserstein GAN),一些新方法被证明在批次效应校正方面有更好的效果;例如,基于从SciBet学习到的带注释数据集中的生物学先验知识,SSBER可以在大型RNA测序数据集中去除批次效应(Zhang and Wang,2021)。建议在整合单细胞转录组数据之前,应根据数据的实际情况先测试多种方法,然后选择最合适的批次效应去除方法。例如,Jinmiao Chen团队和Charles Wang团队分别于2020年和2021年对本综述2.2中提到的前三种方法的大部分进行了基准测试,证明了Harmony和Seurat V3在大多数情况下都能达到良好的批次效应校正效果,这符合这两种方法如今仍然被广泛使用,但对于深度学习方法来说仍然缺乏好的指标这一事实(Chen等,2021a ;Tran等,2020)。
(3)填补
测序过程中会引入大量0值(高通量大规模10x基因组测序数据中零值可能超过90%)(Stegle等,2015 ; Talwar等,2018),这会干扰下游生物学差异分析,因此必须对原始基因表达矩阵中的缺失数据值进行填补,同时有效区分技术噪音零值与生物学零值。
基因 / 细胞分离方法主要应用于早期的填补,其分别考虑细胞相似性(MAGIC ( van Dijk等,2018 )、Sclmpute ( Li and Li, 2018 )、VIPER ( Chen and Zhou, 2018 )、RESCUE ( Tracy等,2019 )、scRMD ( Chen等,2020a )、scRoc ( Ran等,2020 ))或基因间关系(SAVER ( Huang等,2018a )、SAVER-X ( Wang等,2019a )、G253 ( Wu等,2021e )、DCA ( Eraslan等,2019 )、DeepImpute ( Arisdakessian等,2019 ))。总体而言,这些方法缺乏对数据集整体的考虑,容易导致过度插补或者引入误差(Zhang等,2019d)。综合方法综合考虑细胞与基因之间的联系:CMF-Impute和netNMF-sc是最早有效利用细胞与基因之间的关联进行插补的方法(Elyanow等,2020 ;Xu等,2020a)。scIGANs通过特定的GAN模型处理基因表达矩阵,利用生成的细胞训练GANs模型来插补dropout(Xu等,2020b)。近年来,新的方法还在不断被提出,以更好地解决dropout之外的技术噪声对数据的影响,并实现对生物零值的更好的区分。AutoClass(Li等,2022c)实现了无监督处理,而ALRA方法主要针对生物零值(Linderman等,2022)。scMOO进行了根本性的改变,利用数据的潜在结构来学习细胞相似性垂直结构和总低秩结构中的深度关联,从而取得了比单一基因表达矩阵作为输入更好的插值效果,但对内存的要求也更高(Jin等,2022a)。sc-PHENIX利用PCA-UMAP初始化方法,实现了基因表达的非线性插值(Padron-Manrique等,2022),目前哪种插值能取得最佳效果尚无明确定论。由于数据集本身的原因,下游分析的目的会有不同的选择,但毫无疑问最好的填补方法将能够以较低的计算要求有效区分技术噪声零值和生物零值(Jiang等, 2022a ;Wen等, 2022)。
(4)特征选择
为了降低数据维数以提升计算分析效率、减少技术噪声干扰和模型过拟合的风险,我们常常采取特征选择策略,选取不同细胞中差异较大的基因,而非整个数据集基因进行聚类等后续分析(Brennecke等, 2013 ;Jackson and Vogel, 2022 ;Svensson等, 2017)。
在bulk RNA-seq分析中,寻找差异基因的方法通常包括基于倍数变化(FC)的方法、基于统计检验的方法和FC-统计检验方法,显然后者的筛选结果和可信度最好(Chung and Storey,2015)。
早期的单细胞特征选择方法缺乏平均表达量与方差之间的校正,导致结果中高表达基因的比例过高(Brennecke等,2013)。EDGE采用大量弱学习器的集成学习方法来学习细胞间相似性概率,提取基于信息熵的显著贡献作为高可变基因(Sun等,2020c)。同样,SAIC基于迭代聚类最终输出实现了最优细胞簇分离(Yang等,2017)。近期,一些新的特征提取策略被提出并证明了其稳定性和有效性,但它们之间的性能权威验证尚缺乏:包括基于基因表达分布矩阵的方法 SCMER(Liang等,2021b)、RgCop(Lall等,2021)、scPNMF(Song等,2021a)、SIEVE(Zhang等,2021e);基于熵的方法 IEntropy(Li等,2022g)、infohet(Casey等,2023);综合考虑聚类的方法有Triku(Ascensión等,2022)、FEAST(Su等,2021)等。由于上述方法绝大多数忽略了整体的依赖于基因表达的特征,因此提出了综合的方法,如Triku使用k最近邻图的方法对基因表达模式进行综合探索和分类,实现无偏差地筛选出更有生物学意义的特征基因;FEAST在共识聚类上通过f检验对特征进行排序,并基于特征评估算法提取HVG(Wang等,2022c)。
其他一些方法使用高可变基因以外的特征来表示数据集,例如scVEGs和scSensitiveGeneDefine方法,使用高变异系数(CV)作为特征提取;BASiCS方法利用spike-in基因的信息(Chen等,2016b ;Chen等,2021b)。总体来看,基于准确性、生物学可解释性等角度,当前特征选择的主要目标是有效提取HVG,以便对高维转录组数据进行有效的下游分析。
(5)降维
由于单细胞转录组通常包含数万个甚至更多的基因,不利于直接提取有效信息,在实际分析过程中,通常需要对原始测序数据进行降维。除了利用前文提到的特征选择方法处理高维单细胞转录组测序数据外,降维也是一种有效的方法,根据降维策略可分为线性降维(基于潜在狄利克雷分配(LDA)的方法、基于PCA的方法)和非线性降维(基于t-SNE的方法、基于UMAP的方法)(Andrews and Hemberg,2018 ;Becht等,2019 ;Laurens and Hinton,2008 ;Peres-Neto等,2005)。
在线性降维中,LDA和PCA是两种广泛使用的算法,LDA从分类最大的角度区分特征,而PCA则从方差最大的角度正交提取主成分。尽管有JPCDA、LDA-PLS等改进算法,但是LDA模型在单细胞转录组数据中的降维效果仍然不是最优的(Tang等,2014 ; Zhao等,2020)。PCA是另一种线性变换,Seurat通常根据标准差-PC图的拐点或者PC的比例检验结果P值(ScoreJackStraw函数)来确定PC数量的多少。其他基于PCA的降维方法的变体包括pcaReduce(Žurauskien ė和Yau,2016),GLM-PCA(Townes等,2019),RPCA(Gogolewski等,2019),tRPCA(Candès等,2011),scPCA(Boileau等,2020),PCAone(Li等,2022l)。GLM-PCA将传统PCA分析扩展到非正态分布,通过引入指数家族似然策略直接处理原始矩阵,使PCA摆脱正态化限制,然后使用偏差对基因实现进行排序和提取(Collins等,2002)。ScPCA使用对比PCA和稀疏PCA分别去除技术噪音和数据,进一步增加了PCA的稳定性(Abid等,2018 ; Zou等,2006)。由于大多数scRNA-seq数据集难以通过简单的线性降维进行有效表示,解决这一问题的第一个策略是基于快速PCA分析方法。PCAone提出了一种新的快速随机奇异值分解(RSVD)策略,在35分钟内完成130万小鼠脑细胞单细胞数据的分析(Li等,2022l)。
非线性降维是另一种解决方案,如非参数降维方法t-SNE和UMAP,都需要预先设置聚类的超参数;而在分类效果上,前者倾向于离散数据中细胞的形成。在合理使用参数设定的情况下,UMAP与t-SNE并无明显差异,即在使用相同的信息初始化方法后,二者可以在保留数据集全局结构的同时,产生近似的分析效率(Do and Canzar,2021 ;Kobak and Linderman,2021)。针对t-SNE的改进方法包括net-SNE、qSNE、FItSNE、联合t-SNE(Cho等,2018a ;Linderman等,2019 ;Wang等,2022b),而UMAP的改进主要来自于Leland McInnes课题组对该方法的自我改进(McInnes等,2018)。为了更好地可视化t-SNE或UMAP的降维结果,Hyunghoon Cho提出了基于局部半径依赖优化的转录组变异信息den-SNE/densMAP方法,以迭代优化传统t-SNE/UMAP的功能;Stefan Canzar提出了j-SNE/jUMAP来改善多模态组学数据联合可视化结果,减少可视化的误导性(Do and Canzar,2021 ;Narayan等,2021)。
(6)聚类
在单细胞转录组数据分析中,通过聚类将细胞划分为亚群,从而能够表征多细胞生物中不同细胞类型,这有助于我们从细胞异质性的角度准确地分析不同的组织或发育过程。聚类的实际效果会受到数据预处理步骤的影响,例如浴效应归一化、归纳、降维等。
在特征基因选择和降维之后,绝大多数单细胞是基于距离进行聚类的。K 均值聚类算法的概念被用于 SCUBA、SC3 和 RaceID 等应用(Grün等,2015 ;Kiselev等,2017 ;Macqueen, 1967 ;Marco等,2014)。在参数选择改进方面,SAIC 通过 Davies-Bouldin 指数迭代优化多个初始中心K和P值,以获得最优解;LAK 将参数选择算法应用于数据集,实现参数的自动选择(Davies and Bouldin, 1979 ;Hua等,2020 ;Yang等,2017)。在超高维数据的操作中,LAK添加Lasso惩罚项进行标准化,mbkmeans使用小批量k均值实现百万细胞级别的快速聚类(Hicks等,2021)。SMSC应用谱聚类方法来提高聚类性能,但对于超高维数据会损失一定的准确性(Qi等,2021)。另一大类广泛使用的距离聚类方法依赖于共享最近邻图结构和图聚类,其中使用最广泛的是Louvain或Leiden(Blondel等,2008 ;Xu and Su, 2015)。稀有细胞的识别需要结合特定方法进行改进,例如dropClust使用局部敏感哈希工作流筛选最近邻,然后是Louvain聚类,它使用指数衰减函数来保留更多稀有细胞的转录组特征(Sinha等,2018)。其他基于距离的聚类方法使用不同的算法核心:SIMLR使用高斯核学习模型为数据集中潜在的C细胞群体构建核矩阵,而Conos提出联合相互最近邻(mNN)图聚类来实现对多个不同单细胞转录组样本的整合分析(Barkas等,2019 ; Wang等,2017a)。基于密度的聚类利用样本分布的接近程度进行聚类,DBSCAN是最经典的算法(Ester等,1996 ; Fukunaga and Hostetler, 1975)。对于单细胞测序,densityCut和FlowGrid就是基于此原理设计的(Ding等,2016 ; Fang and Ho, 2021)。层次聚类是一种自下而上的聚类方法,通过无监督学习,不断重复计算细胞与细胞的相似性进行分类,直至完成预设的聚类数(Guo等,2015)。随后,RCA聚类采用常规的层次聚类方法,对映射到全局参考面板的细胞进行聚类;HGC在SNN图上构建层次树(Li等,2017 ;Zou等,2021)。为了解决常规层次聚类方法难以对某一组细胞进行聚类、只允许同一组特征基因进行聚类的缺陷,K2Taxonomer采用约束K均值算法扩展到样本组,基于多个基因集递归进行积分计算,以捕获各种分辨率下的亚组(“类似分类学的细胞”)(Reed and Monti, 2021)。Mrtree将层次聚类的策略应用于平面簇的多个划分,并构造多分辨率协调树用于细胞聚类(Peng等,2021a)。最近, Zelig和Kaplan(2020)提出了一种KMD聚类方法,通过平均链接层次聚类模型在聚类时消除了超参数K,大大减少了主观性带来的判断误差。
深度学习聚类方法是将机器学习方法与上述单细胞转录组聚类策略相结合,可以以无监督、监督或半监督的形式实现更高效的聚类结果。这些方法倾向于学习一种非线性变换,通过将原始高维数据映射到较小的潜在空间中来获得最佳的低维表示。总体而言,这种方法避免了传统聚类方法对聚类前数据处理方法选择的影响。无监督聚类方法包括ADClust、DESC、SAUCIE、VAE-SNE等,通常不需要预设聚类个数等参数,以自主学习的方式完成对数据集的分析处理(Amodio等,2019 ;Graving and Couzin,2020 ;Li等,2020c ;Zeng等,2022c)。虽然无监督聚类方法避免了手动输入聚类个数等参数,可以延伸到超高维细胞聚类,但有时利用高质量标注数据集或其他先验知识辅助约束进行监督或半监督聚类,可以实现更为准确的细胞类型分类,提高聚类性能(Bai等,2021)。基于迁移学习的ItClust、基于互监督ZINB自编码器和图神经网络(GNN)的scDSC、基于软K均值卷积自编码器的ScCAE、基于Cramer-World距离最大均值惩罚高斯混合自编码器的SeGMA、基于时间序列聚类网络STCN都是广泛使用的监督聚类(Gan等,2022 ; Hu等,2022a ; Hu等,2020a ; Ma等,2021b ; Smieja等,2021)。此外,Zhang团队(Yang等,2023b)利用分层GAN设计了另一种广泛使用的深度学习方法IMDGC,用于单细胞转录组数据分析,以生成的方式构建细胞嵌入簇。
针对聚类中的特殊情况,设计了有针对性的聚类方法:GiniClust(Jiang等,2016)(更新为GiniClust 3(Dong and Yuan,2020))、MicroCellClust(Gerniers等,2021)用于稀有细胞亚群聚类;EDClust(Wei等,2022)、ENCORE(Song等,2021b)和MLG(Lu等,2021)用于降噪和消除批次效应;ClonoCluster(克隆起源信息)(Richman等,2023)、IsoCell(可变剪接信息)(Liu等,2023)使用附加信息进行聚类。Wu 和 Yang 从特征选择对聚类的影响的角度对聚类方法进行了评估,他们证明更具代表性的特征选择会提高细胞聚类的水平,基于“聚类相似性”的方法(我们综述中提到的大多数基于距离的聚类方法)通常具有广泛的高聚类类型性能;然而,高精度和高运行速度需要根据实际数据集进行有针对性的选择(Su等,2021 ;Yu等,2022)。双重浸入 (double dipping)是一个显著的问题,即在细胞聚类和差异表达基因中使用相同的表达数据,导致在细胞聚类不正确时 DE 基因的错误发现率 (FDR)过高。例如,如果只存在一个特定的细胞聚类,则不应将任何基因视为差异基因。为了系统地解决这个问题,ClusterDE 采用了聚类对比学习策略进行聚类后 DE 测试。该方法与截断正态分布 (TN)检验和 Countsplit 方法相比,在不同阈值范围内具有更好的 FDR 控制 ( Song等,2023a )。
(7) 细胞类型注释
细胞分型注释是指利用特定的信息对单细胞测序数据集中的细胞或细胞亚群进行注释,作为后续生物学分析的基础。最常用的策略是对细胞进行无监督聚类,然后根据标记基因进行注释,例如 scCATCH、SCSA ( Cao等,2020b ;Shao等,2020a ),但它难以处理复杂的高维数据集 ( Franzén等,2019 ;Luecken and Theis, 2019 ;Zhang等,2019c )。目前已经开发了多种自动细胞分型方法,大致可分为两类,即依赖参考和无参考的注释方法。
依赖参考信息的注释方法要求用户提供预先注释的高质量单细胞转录组数据集或来自 PanglaoDB 数据库、ScType 数据库等的先验知识进行比对(Ianevski等,2022)。根据方法原理的不同,可分为基于层次树的方法(CHETAH(de Kanter等,2019)、Garnett(Pliner等,2019)、HieRFIT(Kaymaz等,2021)、scHPL(Michielsen等,2021)、scMRMA(Li等,2022e))、基于相似性的方法(SingleR(Aran等,2019)、scmap(Kiselev等,2018)、deCS(Pei等,2023)、scID(Boufea等,2020)、scMatch(Hou等,2019)、Symphony(Kang等,2021))、基于签名基因的方法(Cellassign(Zhang等,2021))、基于特征基因的方法(Cellassign(Zhang等,2022))。al., 2019a )、Cell-ID(Cortal等,2021)、scMAGIC(Zhang等,2022g)、SciBet(Li等,2020b))和其他DL方法。作为早期方法,ACTINN是一种使用3个隐藏层神经网络进行注释分类的深度学习方法(Ma and Pellegrini, 2020)。SCPred随后提出了一种基于嵌入的无偏特征选择的机器学习概率预测方法(Alquicira-Hernandez等,2019)。其他方法如Seurat在PCA空间中投影查询细胞并通过加权投票分类器训练细胞分型注释;scSorter 采用高斯混合模型,GraphCS 使用虚拟对抗训练 (VAT)损失修改的 GNN 来扩展到多物种、大规模细胞注释数据集(Guo and Li,2021 ;Zeng等,2022a)。
不依赖参考信息的注释方法使用预先训练的深度学习模型,可以直接使用查询数据集作为输入进行细胞分类。scDeepSort 使用来自人类细胞图谱 (HCL)和小鼠细胞图谱 (MCA)数据库的单细胞图谱作为预训练加权 GNN 模型的输入,该模型适用于人类和小鼠细胞注释并取得良好的效果(Han等,2018b ;Han等,2020 ;Shao等,2021b)。类似地,Pollock 是一个预训练的人类癌症参考 VAE 模型,用于对癌症环境中的多模态细胞进行分类(Storrs等,2022)。虽然使用起来更方便,但对于差异显著的查询数据集难以达到更好的细胞注释效果,而且由于准确性和预训练参考数据集的数量也难以扩展应用。还有一些其他用于有针对性领域研究的细胞注释工具,例如,用于人类肾细胞注释的 DevKidCC(Wilson等,2022),用于识别癌症和正常细胞的 ikarus(Dohmen等,2021)。总体而言,无参考注释方法的性能受到预训练参考数据集的覆盖率和准确性的制约。
目前,改进细胞注释工具以在大平台和多细胞模式下统一分配细胞类型是细胞注释研究的主流方向,最新的Cellar和ELeFHAnt方法在这方面做了一些尝试并取得了初步成果(Hasanaj等,2022 ; Thorner等,2021)。总体而言,基于相似性的注释方法计算量大,在应用于非常大的查询和参考数据集时,往往会在准确率和速度之间做出权衡,因此一般只适合在较小的数据集中进行细胞分类;对于较大规模的数据集,建议使用F检验特征选择或MLP分类器(Hu等,2020a ; Huang and Zhang, 2021 ; Ma等,2021c)。此外,半监督迁移学习的方法,如Itclust,在发现新的细胞亚型方面也有不错的效果。近年来,基于上述参考注释方法分类的新方法不断完善,VAE等深度学习模型也在该领域得到应用。
(8)差异表达分析(DEG )
统计检验是Bulk RNA-seq的差异基因分析中常用到的方法,类似章节2.4HVG Selection算法:通常以P值和对数倍变化量作为重要参数。统计检验包括t检验(两个样本为基础),Wilcoxon检验,Kolmogorov-Smirnov检验(KS检验),Kruskal-Wallis检验(KW检验),其中一些在单细胞转录组DEGs的检验中也被广泛使用。基于此,发展了相应的检测工具:limma(Ritchie等,2015),edgeR(Robinson等,2010),DESeq2(Love等,2014)。limma和edgeR算法均由Smyth GK提出,前者基于正态或近似正态分布模型,后者基于过度离散的泊松分布模型。DESeq2基于NB分布模型进行假设检验,对DEG采用经验贝叶斯程序。目前limma由于特定的分布模型假设,在RNA计数分析中误差较大,虽然edgeR和DESeq2都利用贝叶斯模型对过度离散进行归一化,但后者通过数据集reads的平均值和异常值检测促进了CPM阈值的筛选,分析效果更好。
单细胞转录组DEG按照时间和分析方法大致可以分为早期零值参数检验、非参数检验和其他方法。由于scRNA-seq数据中存在大量零数,早期的方法大多基于此观察做参数检验,例如Monocle ( Trapnell等,2014 )、SCDE ( Kharchenko等,2014 )、MAST ( Finak等,2015 )、scDD ( Korthauer等,2016 )、D3E ( Delmans and Hemberg, 2016 )、TASC ( Jia等,2017 )、DEsingle ( Miao等,2018 )和HIPPO ( Kim等,2020b )。对以上一些方法的评测表明,虽然它们在单细胞数据集的分析中普遍取得了不错的效果,但对于批量数据(Soneson and Robinson, 2018)相比DEA方法并没有明显的性能提升。对于不同的数据集,有可能没有最好的分布模型,因此一种替代解决方案是考虑非参数DEA方法。
非参数检验或无分布检验不需要对数据分布形式做事先假设,因此适用于多数据集的分析,常用方法有Swish(Zhu等,2019a)、IDEAS(Zhang等,2022d)、ccdf(Gauthier等,2021)、distinct(Tiberi等,2022)。Swish通过Salmon Gibbs评估转录本水平,然后用Mann-Whitney Wilcoxon检验计算FC值。IDEAS是一种使用Jensen-Shannon散度(JSD)或Wasserstein距离(Was)进行基因差异表达测量的伪F统计量检验, P值由基于PERMANOVA的距离测试器基于核的回归生成。Ccdf 是一种依赖条件累积分布函数的条件独立性检验,通过多元回归模型预测 DEG。Distinct 提出了一种分层非参数置换方法,使用经验累积分布函数 (ECDF)的总距离进行 DEG 识别。替代方法包括深度学习策略 MRFscRNAseq ( Li等,2021a )、基于拟时序推断的 PseudotimeDE ( Song and Li, 2021 )、基于非预聚类的 singleCellHaystack ( Vandenbon and Diez, 2020 )、基于多重评分的 MarcoPolo ( Kim等,2022 )。建议不同的单细胞转录组数据集应采用数据特定的DEGs检测策略,以优化DEGs分析,基于scCODE工作流程,可以使用涉及CDO(DE基因顺序)和AUCC(一致性曲线下面积)的指标找到最优化的DEGs方法(Zou等,2022)。此外,研究方法在不同的研究背景下会有特定的研究取向,例如在给药后的剂量反应研究中,DEGs分析、LRT线性检验和贝叶斯多组检验均比其他方法有更好的结果(Nault等,2022)。
(9)可视化
单细胞转录组数据分析可视化是指将上述分析结果以图形的形式直观地呈现,ggplot2是R中最广泛的可视化工具,在R中被广泛使用,可以大大增强绘图能力(Wickham,2009)。ARL 是另一个专门显示标记基因关联图并可显示其在每个簇中的特征的 R 包(Gralinska等,2022)。此外,还有其他专门用于标记基因可视化的包,如 Complex Heatmap,本文不再详细介绍。HVG 可视化通常以火山图的形式呈现,默认情况下,图的左侧和右侧部分分别是代表性不足的基因和代表性过高的基因,而中间是恒定基因。Enhanced Volcano 是一个专门用于绘制火山图的 R 包,默认情况下也可以使用 ggplot2 来获得更好的结果。簇可视化通常以 PCA 图、t-SNE 图和 UMAP 图呈现,但值得注意的是,可视化的结果非常具有欺骗性,因为一些小的细胞亚群可能代表 UMAP 图中显示的大量细胞。为了解决这些问题,提出了den-SNE/densMAP、j-SNE/j-UMAP等改进方法(Macqueen,1967 ;Marco等,2014)。此外,FastProject可以输出注释簇的2D显示,PieParty可以在簇2D图中为每个基因绘制颜色图(DeTomaso and Yosef,2016 ;Kurtenbach等,2021)。
同时,单细胞转录组数据的交互式可视化是目前的热门领域,诸如Single Cell Explorer等软件可以一定程度上实现交互式可视化,但仍需增加交互自由度,以提供更全面的单细胞转录组数据3D呈现(Cakir等,2020 ;Feng等,2019)。为此,CellexalVR利用VR理论进行交互可视化;CellView 是一个基于 Web 的工具,包括用于不同用途的探索选项卡、共表达选项卡、子簇分析选项卡模块;Cellxgene VIP 是一个基于 cellxgene 框架的插件,并扩展到基于多个模块组合的 ST 数据的交互式可视化(Bolisetty等,2017 ; Legetth等,2021 ; Li等,2022f)。
(10)单细胞模拟
随着单细胞转录组方法的不断扩展,基准测试成为了重要挑战,关键问题是需要稳定可靠的数据,因为单细胞转录组的直接测序可能缺乏基本事实。真实的单细胞模拟数据为基准测试提供了已知的事实,允许使用真实数据进行训练,同时匹配实际数据的特征。此外,模拟数据比真实数据提供了更大的灵活性,使分析师能够根据特定的测试方法调整诸如 dropout rate 等参数。
Splatter 是一个两步模拟框架,首先模拟来自真实数据的估计参数,然后合并来自用户的额外参数(Zappia 等,2017)。其六个预先设计的管道模块接口确保了数据生成的可重复性。最近的更新侧重于专业化和泛化。在专业化领域,splaPop 生成具有遗传效应(数量性状基因座)的人口规模数据,而 dyngen 模拟动态细胞过程,如发育轨迹(Azodi 等,2021 ;Cannoodt 等,2021)。在泛化领域,Li的团队介绍了理想模拟的六个概念,包括真实性、基因的保存、基因相关性的捕获、稳健性、参数可调性和效率(Song 等,2023b ;Sun 等,2021)。随后,scDesign2 提出来满足所有 6 个属性(Sun等,2021),接着是 scDesign3,解决单细胞组学统计模拟的空白(Song等,2023b)。模拟准确性和透明度的提高增强了不同单细胞数据处理方法之间的基准测试,指导选择最合适的方法以满足特定数据和许可需求。
下游拓展分析
(1) 拟时序分析
为了更真实地恢复生物体中的真实过程,需要使用拟时序分析整合多个转录组数据,通过推断不同时间点的细胞信息(包括状态、分布、数量和基因表达)来重建细胞发育轨迹(BarJoseph等,2012 ; Bendall等,2014 ; Ding等,2022)。这种对转录组特征的动态分析称为拟时序分析(图 4 E)。根据是否依赖于基因表达,拟时序分析方法可以分为基于基因(外显子)表达的方法和基于RNA-velocity的方法。
基于基因表达水平的拟时序分析最早被提出,它通常利用降维等聚类方法在低维空间构建多分支图模型来模拟细胞的发育轨迹:基于最小生成树(MST)的方法monocle( Trapnell等,2014 )、monocle 2( Qiu等,2017 )、TSCAN( Ji and Ji, 2016 );基于PAGA 的方法PAGA( Wolf等,2019 )、monocle 3( Cao等,2019 );其他图架构方法Wishbone( Setty等,2016 )、p-Creode( Herring等,2018 )等都用于此目的。MST是连接二维平面上所有点的模型,具有最低的总连接权重,最早用于解决旅行商问题, Qiu et al. (2011)在2011年应用Boruvka算法构建的MST模型来分析细胞层级。Monocle将细胞映射到高维欧氏空间,并使用ICA降维,Monocle 2更新单片机并使用反向图嵌入(RGE)策略构建细胞路径,随后细胞分布到使用质心构建的生成树上。PAGA(基于分区的图抽象)通过邻域图权重(默认为KNN图)的统计连通性度量保留数据集的全局拓扑结构,基于扩展扩散拟时序(DPT)方法生成多种分辨率的PAGA图进行拟时序分析。Monocle 3结合了monocle 2和PAGA的优点,在UMAP空间上形成多个PAGA图,然后使用SimplePPT算法学习主图,再通过其他PAGA图的约束,最终得出的细胞发育轨迹可以适应具有成分复杂性的大数据集。总体而言,PAGA和monocle 3综合考虑了计算速度、准确性和鲁棒性,是目前单细胞转录组拟时序分析的最佳方法。除了图方法外,其他基于基因表达的方法还包括CSHMM,利用HMM模型计算每个细胞到根细胞的距离,然后迭代完成细胞轨迹分配;SCUBA,采用分叉分析模型;SLICE,由于高度分化的细胞使scEntropy最小化,因此提出了一个scEntropy有向模型(Guo等,2017 ;Lin and Bar-Joseph,2019 ;Marco等,2014)。
基于RNA速率的方法依赖于RNA速率信息,该方法由Peter V. Kharchenko小组(La Manno等,2018)于2018年首次提出,他们认为未剪接/剪接mRNA的比例可用于推断转录动力学,因为未剪接mRNA比例较高的细胞更年轻(作为较晚的细胞分化状态)。同时,他们还提出了专门的分析软件velocyto(可通过R包velocyto.R获得)作为稳态模型来量化RNA速率以进行发育轨迹分析。scVelo是另一种专门为RNA速率设计的分析工具,它使用基于似然的动力学模型来解决具有稳态mRNA水平的细胞轨迹推断,并且情况违反了常见剪接速率的中心假设(Bergen等,2020)。但速度投影方法仍有方法论改进的空间:恒定降解和核输出假设仍需证明。这也为后续基于RNA速率的方法提供了方向(Bergen等,2021)。深度学习相关方法被广泛应用于RNA速率的建模预测,以进一步增强对大型复杂数据集的处理能力,如贝叶斯分层模型BRIE2(Huang and Sanguinetti,2021);基于速度自编码模型的VeloAE(Qiao and Huang,2021);变分自编码模型DeepCycle(Riba等,2022)。
(2)细胞间相互作用
细胞间相互作用(CCI)是多细胞生物维持正常生理功能的重要特征,决定了细胞的命运,探讨疾病发生的机制,探索遗传变异过程和其他调控过程(Shao等,2020b ;Singer, 1992)。细胞相互作用网络直观地体现了细胞间的相互作用关系(图4 F)。
基于邻域结构的直接CCI是指利用细胞间的物理距离,对有可能直接接触的CCI进行提取和分析。ProximID方法对具有预定相互作用距离(欧几里德距离)的合格细胞完成物理细胞网络构建(Boisset等,2018)。Neighbor-seq使用随机森林分类器识别细胞类型,通过igraph方法利用富集分数计算得分构建CCI网络(Csardi and Nepusz, 2006 ; Ghaddar and De, 2022)。由于这种分析方法的局限性很大,目前并不单独使用。这种KNN连通图通常作为深度学习中CCI的输入之一,其物理距离也成为单细胞CCI研究中的一个重要假设(两个物理上直接接触的相邻细胞比两个随机细胞更有可能发生某种形式的相互作用),用于全局CCI分析。
间接接触的CCI关系完整过程应包括配体、受体、信号蛋白、转录因子(TF)和靶基因。常见的间接CCI方法主要利用先验的配体-受体对数据库(如CellTalkDB数据库,该数据库整合了经过验证的3,398个人类LR对和2,033个小鼠LR对的信息(Shao等,2021a)),制作细胞-细胞连接矩阵,其中每个值代表LR对的共表达水平。然后构建细胞连接图进行CCI分析,主要的分析方法包包括单细胞CCI推断方法SoptSC(Wang等,2019d)、Scriabi(Wilk等,2024);基于LR对的集群CCI方法NATMI(Hou等人,2020)、SingleCellSignalR(Cabello-Aguilar等人,2020)、scCrossTalk(Shao等人,2024)、CellPhoneDB(Efremova等人,2020)、Nichenet(Browaeys等人,2020)、CellChat(Jin等人) al., 2021)、CellCall(Zhang 等人,2021d)、ICELLNET(Noël 等人,2021)、 scMLnet(Cheng 等人,2021)、CytoTalk(Hu 等人,2021b)、Tensor-cell2cell(Armingol 等人,2022)、LRLoop(Xin 等人) 2022);其他基于信息的聚类CCI方法有InterCellDB(Jin等,2022b)、EBOCOST(Zheng等,2022b)。基于LR对的方法使用文献数据库整理或之前自我验证的LR对构建数据库:NATMI默认使用connectomeDB2020数据库(2,293个LR对中有1,751个来自作者在2015年验证的草图)来构建加权有向多边网络(Ramilowski等,2015)。CellPhoneDB提出了一种特定的SQLite数据库来保留LR对的特定亚基架构,使用平均表达阈值来确定相互作用的细胞,并使用几何草图子样本框架来增强对大数据集的功效并排除噪音。同样,ICELLNET利用异源复合物中配体与受体的多亚基结构。NicheNet 在LR 先验模型上采用基于模型的参数优化,通过添加细胞内信号信息(靶基因)来优化 CCI 强度,克服了上述方法直接用受体基因表达水平表示细胞内受体蛋白数量以及结合下游信号通路与 GRN 改进 CCI 分析的问题。因此在单细胞转录组 CCI 分析中,通常将 CellPhoneDB 与 NicheNet 结合使用以达到最佳分析效果(Dimitrov等,2022)。
最新的单细胞 CCI 方法采用了 DL 的策略,在一定程度上提高了应用性能。DeepLinc 使用 VGAE 模型重建全范围细胞间 CCI 网络(Li and Yang, 2022)。TraSig 是一个连续状态隐马尔可夫模型,使用拟时序排序计算动态交互分数进行 CCI 推断(Li等,2022a)。此外,由于现在空间分辨的转录组学 (ST)为基因信息提供了至关重要的空间信息,推断空间细胞间通讯仍然是一个巨大的挑战。SpaOTsc 可以重建 scRNA-seq 数据的空间属性,并依靠结构化的最佳传输方法构建 CCI 网络 ( Cang and Nie, 2020 )。Giotto 使用细胞间邻近图来推断信号通路 ( Dries等,2021b )。然而,SpaOTsc 和 Giotto 都很难解析基于点的 ST 数据。最近,范实验室( Shao等,2022a )提出了 SpaTalk,它使用知识图和图网络为单细胞和基于点的 ST 数据构建空间相邻细胞之间的配体-受体-靶标网络。
(3)通路富集分析
基因通路富集分析是指以感兴趣的基因作为前景基因与已知的特定数据库关联,建立基因-生物过程链路,用于解释差异表达基因的生理功能、上下游通路等(Creixell等,2015)。基因本体论(GO)和京都基因与基因组百科全书(KEGG)是首批提出用作富集分析的数据库(Ashburner等,2000 ;Ogata等,1999)。基因集富集分析(GSEA)是另一种广泛使用的方法,通过计算富集得分来确定基因集S是否会在ranker DEGs列表L的两侧出现,以及显著性检验值(Subramanian等,2005)。Ingenuity 通路 Analysis (IPA)软件中所有通路都经过实验验证,与其他分析相比,它可以预测激活时整个通路的变化趋势。其他常见的数据库通路富集方法还包括过度表达分析(ORA)(Khatri等,2012)、基于网络拓扑的分析(NTA)(Wang等,2013)、Reactome基因集(Fabregat等,2018)、CORUM复合体(Ruepp等,2010)。参考数据集的来源通路和富集手段的差异会直接影响通路富集结果。为了方便分析,已经提出了集成不同数据库的基于Web的在线分析工具(Wang等,2017b ;Zhang等,2005)。Metascape涉及转录组数据库(KEGG、GO、CORUM、TRRUST等)和蛋白质-蛋白质相互作用数据库(STRING、BioGrid、OmniPath等),共有25个数据库可用于人类和小鼠等8个物种的遗传和蛋白质组富集分析(Zhou等,2019b)。
综上所述,虽然我们列举了单细胞转录组下游分析最常见的部分(表 S3和S4),但仍然有很多方法没有涉及到,包括基因调控网络分析、免疫分析、细胞周期分配、基因变异探索、可变剪接分析等。总体而言,单细胞转录组分析方法多种多样且仍在不断发展,所有分析方法的出发点和最终目的都是利用从单细胞转录组测序数据中精准挖掘出的生物学信息进行生物学解释。
参考文献:
Sun F, Li H, Sun D, et al. Single-cell omics: experimental workflow, data analyses and applications. Sci China Life Sci. 2025;68(1):5-102. doi:10.1007/s11427-023-2561-0